In this exercise, please use Mathematica, which you can download Mathematica from here. NOTE: To download software for which BC has a site license, if you are not physically on campus (hence recognized as being a member of Boston College), then you MUST use the Boston College virtual private network (VPN) -- i.e. use Cisco connect to enable your computer to be recognized as being associated with Boston College.
You'll find the following demo very useful: biostatsBoxplotSummary.nb (PDF file is here). Please download the data Bear Weights, consisting of weights of a collection of bears.
- Using Mathematica, please create a histogram for the bear weights.
-
Please create a relative frequency histogram for the bear weights.
(To do this, use the function
Histogram[data,bspec,hspec]
where "bspec" stands for "bin specification" and "hspec" for "height specification". For "bspec", you can write (for instance) "{min,max,step}", where "min" is the minimum, "max" is the maximum value, and "step" is the distance between one bin and the following bin. Thus "{1,200,5}" gives bins containing values in each of the successive intervals: [1-5], [5-10], etc. To obtain a relative frequency histogram, the argument "hspec" should be either "Probability" or "PDF" (which latter stands for probability density function). - Please create a box wiskers plot for the bear weights.
- Using Mathematics, please compute the number of bears, the arithmetic mean, 10% trimmed mean, geometric mean, harmonic mean, median, sample variance, sample standard deviation, population variance, population standard deviation, first, second and third quartile (the second quartile is the median), range (range is the maximum minus the minimum), and midrange (midrange is the average of the maximum and minimum) values for the data.
Solution: mathematicaSol1.pdf
Solution:
- DNA has bases A,C,G,T while RNA has bases A,C,G,U. It is believed that in a primordial "RNA world", RNA played both an information carrying role (now taken over by DNA) as well as a catalytic role (in many, but not all cases taken over by proteins). DNA is a more reliable information carrier because of DNA repair mechanisms, including SOS. In contrast, RNA suffers from the problem that methylated cytosine spontaniously deaminates and forms uracil. Since uracil is not a valid DNA base, this error can be corrected in DNA though not in RNA. To the best of my knowledge, there is no known error correction mechanism for RNA.
- RNA has a hydroxyl group on the 2' carbon of the pentose sugar, in contrast to DNA which has hydrogen in place of the OH group. This difference allows RNA to form many more hydrogen bonds than DNA; in particular, RNA is generally a single-stranded structure, which forms H-bonds with itself in stable secondary structures.
Solution: 5'-UACGAUCC-3'.
Solution:
- A TATA box is a DNA sequence located 25-35 base pairs upstream of the transcription start site in eukaryotes. Transcription factors (TFs) bind to the TATA box, which then recruit RNA polymerase, which then transcribes mRNA from the gene. The consensus sequence for TATA boxes is TATAAA. Note that only eukaryotes and archaea contain a TATA box, while most prokaryotes contain a Pribnow box, thought to be function as a TATA box, which usually consists of the nucleotides TATAAT.
-
If genes were transcribed from only one strand of DNA, then the answer would
be 3000000000/(4**6) = 732421.875; however, taking into account both the +
and - strands, the answer is
be 6000000000/(4**6) = 1464843.75. Being meticulous, we cannot include the
last 5 nucleotides, so one can argue that the "true" answer is
600000000.-10)/(4**6) = 146484.372559. But even this doesn't account for
the fact that the 3 billion base pairs of double-stranded DNA lie on
46 linear chromosomes (23 pairs of double-stranded chromosomes). So to
be meticulous, one would need make separate computations for each chromosome,
where the plus and minus strands are taken into account of, and the last 5
nucleotides of each strand of each chromosome must be ignored.
Moreover, so far, our computation only concerned the
haploid genome, so if we account for the diploid genome, we must
double the final answer. For the answer to this question, I'm looking for
your reasoning process, not the exact answer, which as we see involves
multiple considerations.
However, the numerical differences between the values when
accounting for the end effect or not are "teeninsy", so one should
not account for end effects in this case. Moreover, we have assumed that
the proportion of each nucleotide A,C,G,T is 0.25 in the human genome. This
is a technically incorrect assumption. If we search for
H. sapiens chromosome 22, complete sequence (since chromosome 22 is
one of the smallest of the 23 pairs of chromosomes)
at the European Nucleotide Archive
of the EBI, we find
files in the primary assembly, one of which has the EMBL accession code
AC005301.22.
Immediately before the nucleotide sequence appears, we find that
Sequence 113688 BP; 37158 A; 22385 C; 21889 G; 32256 T; 0 other
from which we determine thata/bp = 0.32684188304834283
Following convention, the values should be rounded, for instance to the second decimal place yielding
c/bp = 0.19253571177257053
g/bp = 0.19253571177257053
t/bp = 0.28372387587080433a/bp = 0.33
If you now do a more correct computation for the probability of TATAAA, you find a probability of (0.33)4 * (0.28)2 = 0.09025921, which is considerably different than 0.256 = 0.000244140625! (This of course assumes that the compositional frequency in the entire genome is the same as this fragment of chromosome 22.) Note that Chargoff's second "law" (not really a law) approximatly holds.
c/bp = 0.19
g/bp = 0.19
t/bp = 0.28In any case, this is how you go about performing a "Milchmädchenrechnung" (milk maid's computation, the German expression for "back of the envelope computation").
Solution: Search/Analysis -> Peptide Match. Then enter "Homo sapiens" for organism, and use the default settings to get the following:
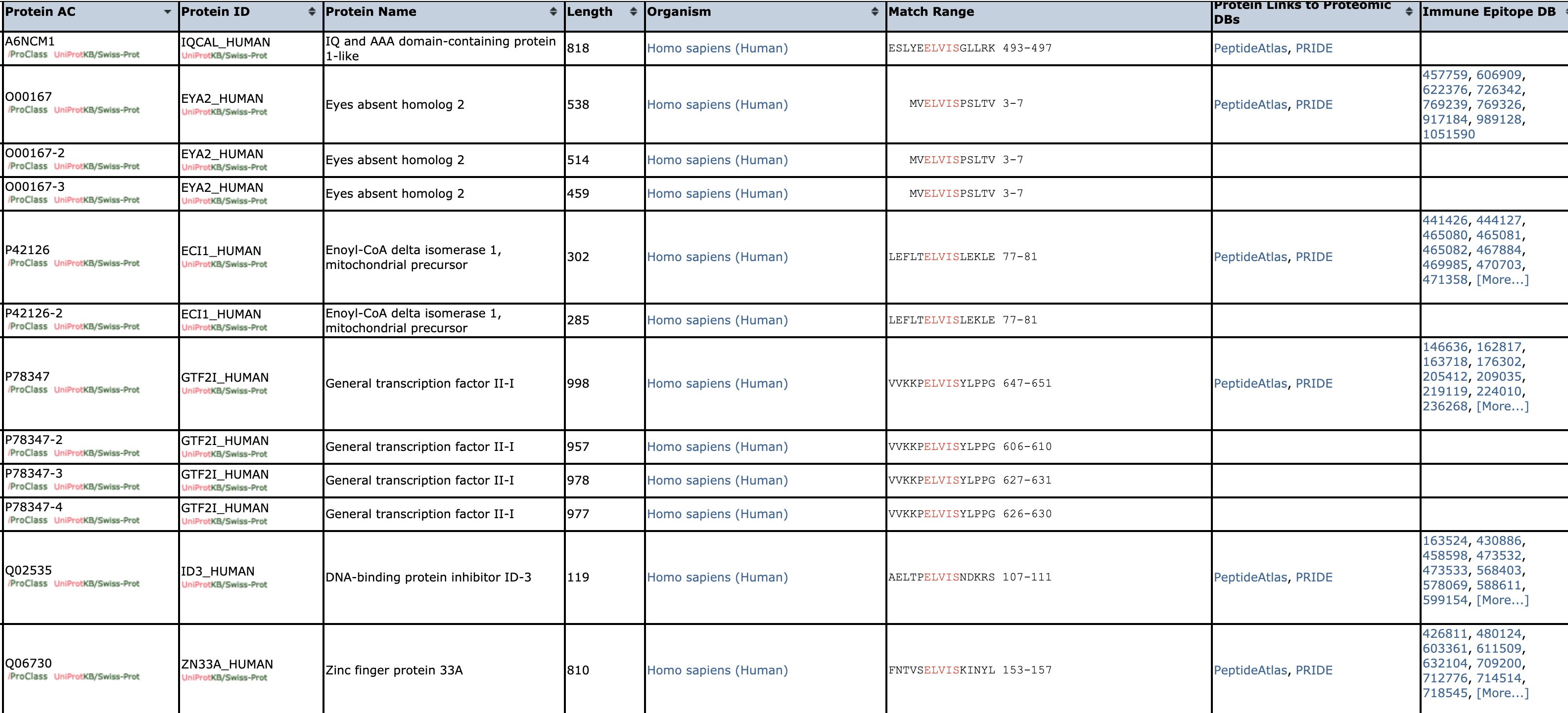
Notice that many of the proteins that contain ELVIS are transcription factors and zinc fingers, both classes of protein that bind DNA. This raises the question of whether ELVIS is a motif for binding DNA -- you would have to do additional research to determine whether this is the case.
Now, if you hadn't first selected "Search analysis -> peptide match", and just entered ELVIS in the search box at the top right, then you get files that simply contain "ELVIS" somewhere in the file, as the examples below indicate.
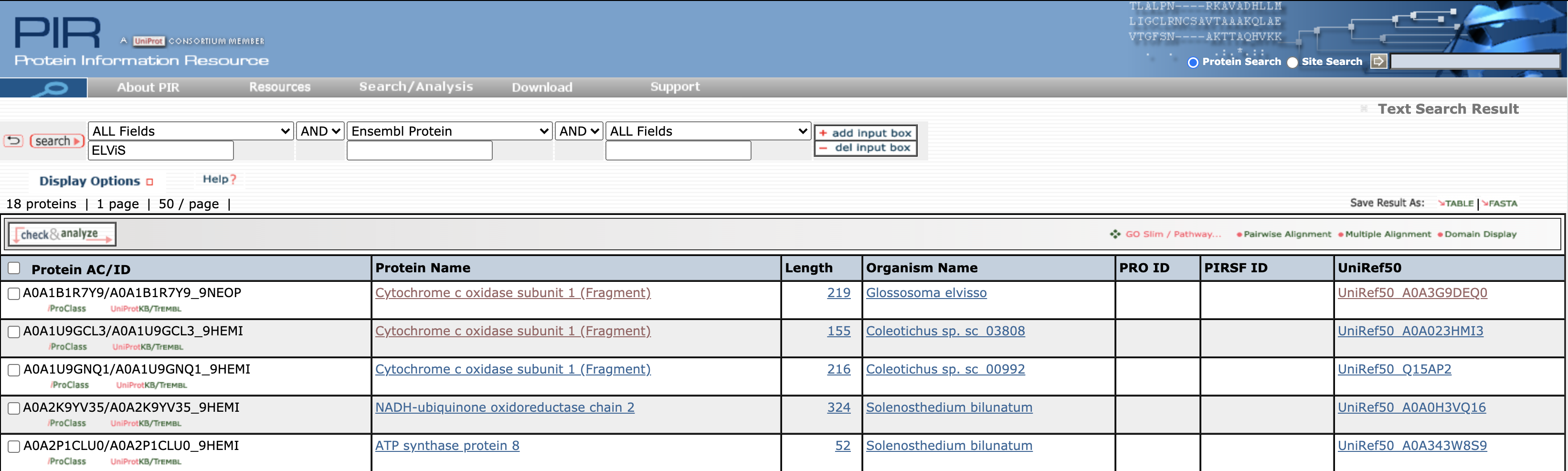
where the only occurrences of ELVIS are in the non-sequence data, for instance, in the first hit, one can look at the GenPept entry for
cytochrome oxidase subunit 1, partial (mitochondrion) [Glossosoma elvisso]where ELVIS is a portion of the organism name. This appears in several similar places in the file, such as in "mitochondrion Glossosoma elvisso", but NOT in the amino acid sequence of the protein.
If instead you use NCBI BLASTp (protein blast), then you find the following:

The above search was not restricted in BLAST to H. sapiens. By restricting to H. sapiens, I found different output, which included, for instance a hit with link to the GenPept file XP_024308862.1 for titin isoform X2 [Homo sapiens]:
14161 lsayaelvis psersdkgiy tlklenrvkt isgeidvnvi arpsapkelk fgditkdsvhTitin (also called connectin) has primary function to give elastic stabilization in myosin and actin filaments.
Solution:
- Isoleucine (IUPAC code I) is similar to leucine (IUPAC code L), as both are hydrophobic and have BLOSUM62 similarity 2.
- Glutamic acid (IUPAC code E) is similar to aspartic acid (IUPAC code D), as both are negatively charged and have BLOSUM62 similarity of 2.
- Serine (IUPAC code S) is similar to threonine (IUPAC code T), as both are small polar amino acids and have BLOSUM62 similarity of 1.
- Histidine (IUPAC code H) is different from leucine, since histidine is polar or hydrophilic, while leucine is hydrophobic. Their BLOSUM62 similarity is -3.
- Cysteine (IUPAC code C) is different from glutamic acid (IUPAC code E), since cysteine is neutral, while glutamic acid is negatively charged, and their BLOSUM62 similarity is -4.
- Tryptophan (IUPAC code W) is different from threonine, since tryptophan is hydrophobic while threonine is polar, and their BLOSUM62 similarity is -2.
Solution: The expected number of random hybridizations (Watson-Crick reverse complements) of an n-length antisense sequence in the 6 billion nucleotides of the human genome is 0.25n * 6 * 109. For this expression to be less than 1, by taking natural logarithms, we need that n*ln(1/4)+ln(6)+9*ln(10) < 0. Algebra yields that n must be greater than (ln(6)+9*ln(10))/ln(4) = 16.24. So choose n to be 17.
Hint: The latter information is not in the GenBank file; however, it IS
in the EMBL file. Go to the EMBL (EBI) server, and search for the GenBank
accession number, click the link "Text" to see the EMBL formatted file,
and look at the line that begins with "SQ" (sequence information) immediately
before the nucleotide sequence of the HIV-1 genome. Later we will learn how
to write a Python program to compute the number of each
nucleotide A,C,G,T.
Solution: Information can be found at
EBI.
The acronym `gag', which stands for `group-specific antigen', refers to
a coding region in retroviruses. In
HIV-1, gag is the gene in the 9238 nt viral genome
that codes for the core structural proteins. The translated gag protein has
amino acid sequence given by:
The genome size and compositional frequency is given byMGARASILSGGELDQWEKIRLRPGGKKKYRLKHLVWASRELERFA VNPGLLETSEGCRQILGQLQPSLQTGSEELKSLFNAVAVLYCVHQRIEIKDTKEALEKI EEEQSKSKKKAQQATADTGSSSQVSQNYPIVQNLQGQMVHQPISPRTLNARVKVIEEKA FSPEVIPMFSALSEGATPQDLNTMLNTVGGHQAAMQMLKETINEEAADWDRLHPVHAGP IAPGQMREPRGSDIAGTTSTLQEQIGWMTNNPPIPVGEIYKRWIILGLNKIVRMYSPAS ILDIRQGPKEPFRDYVDRFYKTLRAEQASQEVKNWMTETLLVQNANPDCKTILKALGPG ATLEEMMTACQGVGGPSHKARVLAEAMSQATNSATIMMQRGNFRNQRRTVKCFNCGKEG HIAKNCRAPRKKGCWKCGKEGHQMKDCTERQANFLGKIWPSHKGRPGNFLQSRPEPSAP PEESFRFGEETTTPSQKQEPIDKELYPLASLRSLFGNDPSSQ
Sequence 9238 BP; 3301 A; 1640 C; 2241 G; 2056 T; 0 other;
Solution:
Fmean 33.225 Fstdev 12.45399225 Mmean 35.475 Mstdev 13.92652422 null hypothesis H0: Fmean=Mmean alternative hypothesis H1: Fmean≠Mmean 2-tailed p-value is 0.44858032, as computed by T.TEST(Fage,Mage,2,3) p-value is NOT < 0.05 (or 0.01) so we can NOT reject the null hypothesis that women's age equal